


Oncology
Lung Synovialosarcoma with Intracanal Infiltration: Case Report and Literature Review
Badache K.1, Habchi N. 1, Djaafer M. 1, Meskouri K.2,
Medjdoub Y.2, Kara Z. 3, Bendjoudi K.4
1: Neurosurgery Department, Mustapha BACHA University Hospital, Algiers, Algeria
2: Thoracic Surgery Department, Mustapha BACHA University Hospital, Algiers, Algeria
3: Orthopedic Surgery and Traumatology department, Mustapha BACHA University Hospital, Algiers, Algeria
4: Resuscitation Department, Mustapha BACHA University Hospital, Algiers, Algeria
Abstract
Synovialosarcoma is a rare malignant tumour, accounting for 10% of soft tissue sarcomas. It usually develops in the limbs and its pulmonary location is exceptional. We report a clinical case of lung synovialosarcoma in a 67-year-old man discovered at a localized stage. This extremely rare tumour, presents an intracanal extension and a wide base of pleural implantation. Multidisciplinary collaboration was ensured: the patient was operated on in two stages, two days apart. The particular immunohistochemical phenotype strongly contributes to the diagnosis. Through this observation, we emphasize the anatomo-clinical, therapeutic and prognostic characteristics of this rare tumor often overlooked by clinicians. We demonstrate the interest of a multidisciplinary collaboration in the management of this tumour.
Keywords: synovialosarcoma, intracanal extension
Introduction
Soft tissue sarcomas are rare tumours, characterized by great anatomical, histological and prognostic heterogeneity. The general incidence of soft tissue sarcomas is estimated between 3 and 4 per 100,000 inhabitants and 50 to 60% of these cancers develop in the limbs [1]. Primary thoracic sarcomas are rare and represent less than 1% of all primary thoracic tumors [2]. Synovialosarcoma is the 4th soft tissue sarcoma (8-10%) in terms of frequency; it affects young adults (15-40 years old) with a slight male predominance [3]. About thirty cases of synovialosarcoma of primary pulmonary site are described in the literature. We report a new observation by presenting the particularities of this tumor rarely encountered in clinical practice.
Patient and observation
Mr. Z.S is a 67-year-old patient, chronic smoker at 25 pack-years and weaned, consulted in March 2022 for chest pain and dyspnea associated with a cough producing hemoptotic sputum in the context of impaired general condition. The physical examination was unremarkable. Furthermore, a notion of tuberculosis contagion was not retained. A thoraco-abdomino-pelvic scan was requested and done on April 14, 2022 showed a voluminous left apico_dorsal tissue lesion of (13 x 11.5 x 18.5 cm) with osteolysis of the vertebral body of the posterior arch of T6 and intra- channel and a broad base of pleural implantation with low abundance left pleural effusion. The abdominal floor showed no abnormality (Figure 1). The patient underwent a CT-guided biopsy, confirming a synovial sarcoma.
A 2nd preoperative thoraco-abdomino-pelvic scan was done on July 24, 2022 with stable aspects compared to the examination of April 14, 2022.

Figure 1: CT images showing left lung invasion of synovialosarcoma with intracanal extension and a wide base of pleural implantation with low abundance left pleural effusion

Figure 2: Aponeuro-muscular disinsertion (A) + laminectomy on two levels D4 and D5 (B) + release of the spinal cord without opening the dura mater (The spinal cord is pushed back by the tumor) + a foraminotomy on both sides (C)

Figure 3 : osteosynthesis of the two levels above and below the lesion with fixation using osteosynthesis material
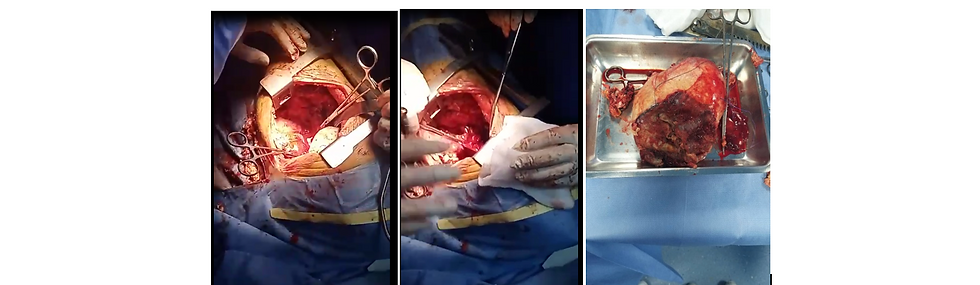
Figure 4 : the posterolateral intercostal approach on the left side, surgical excision of the en bloc expansive tumor process (tumor piece)

Figure 5: Morphological examination of monophasic synovialosarcoma (fusiform cells): microscopic examination of the tumor (HES, magnification x40) Cytokeratin positivity on immunocytochemistry
Multidisciplinary collaboration was ensured:
The patient was operated on in two stages (October 9 and 11, 2022):
The neurosurgeon performed a laminectomy on two levels D4 and D5, we noted that the process pushed back the spinal cord extramedullarily on the right, the release of the spinal cord was performed without opening the dura mater, with a foraminotomy of both listed. (Figure 2).
The orthopedist performed osteosynthesis of the two levels above and below the lesion with fixation using osteosynthesis material. (Figure 3).
The second stage was performed two days after the first surgical stage, the thoracic surgeon approaches the tumor by the posterolateral approach between the ribs, the surgical excision of the expansive process was en bloc, the pulmonary artery was not infiltrated (Figure 4).
The pathological examination of the surgical specimen was in favor of a grade II spindle cell sarcoma (synovialosarcoma) classified as pT3N0Mx with healthy surgical limits without vascular emboli or perineural sheaths. (Figure 5).
Discussion
Soft tissue sarcomas are rare tumours, characterized by great anatomical, histological and prognostic heterogeneity. The incidence of soft tissue sarcomas is estimated between 3 and 4 per 100,000 inhabitants and 50 to 60% of these cancers develop in the limbs [1]. Primary thoracic sarcomas are rare and represent less than 1% of all primary thoracic tumors [2]. Synovialosarcoma is the 4th soft tissue sarcoma (8-10%) in terms of frequency; it affects young adults (15-40 years old) with a slight male predominance [3]. The age of our patient is 67 years old, therefore far higher than the data in the literature, but joins the series of Mastroianni et al whose average age of the patients was between 50 and 70 years old [4]. Most sarcomas have not been associated with risk factors, but certain genetic and environmental predispositions have been suggested in a minority of patients [5].
Clinically, patients with synovialosarcoma of the lung usually present for chest pain, cough; hemoptysis and progressively worsening dyspnea over several months [6]. Our patient's picture is typical of this description. Radiologically, a mass containing calcifications is found in 25% of cases. Computed tomography makes it possible to better appreciate the presence of micro-calcifications as well as the endothoracic and parietal extension. On magnetic resonance imaging, approximately 90% of synovialosarcomas are well circumscribed with a capsule appearance; the presence of lobulations or septa is common. Tumors are heterogeneous on T2 with fluid, solid or fibrous tone signals. The usefulness of positron emission tomography (PET-CT) has been little studied and bone scintigraphy is recommended in case of bone tender point [6].
In the absence of metastases, surgery remains the treatment of choice and wide resection is imperative to reduce the risk of loco-regional and distant recurrences. The interest of adjuvant radiotherapy is to allow better local control of the tumor [ 7]. It is indicated when the tumor has a diameter greater than or equal to 5cm and incomplete margins. No study has assessed the benefit of adjuvant chemotherapy in this situation [7]. Treatment with Doxorubicin and/or Ifosfamide constitutes the first-line treatment in inoperable forms and in metastatic forms with a response rate of around 50% [7]. The average rate of loco-regional or metastatic recurrence at two years is 50% [8]. The most common metastatic sites are lymph nodes, bone and liver. A tumor diameter of less than 5cm, a low mitotic index (Ki 67 < 10%), the absence of tumor necrosis, the absence of residual tumor after surgical resection are considered to be good prognosis factors. Five-year survival varies between 35% and 76% depending on the absence or presence of good prognostic factors respectively [8]. The role of the fusion transcript as a prognostic factor has not been definitively established [8]. In our observation, the patient had a tumor size of 13 x 11.5 x 18.5 cm and a grade II. The tumor size and the absence of adjuvant radiotherapy can lead to early recurrence and unfavorable evolution of the disease.
Conclusion
Lung synovialosarcoma is a rare malignant tumor. Its diagnosis is difficult and requires immunohistochemical and cytogenetic analysis which allows it to be distinguished from other mesenchymal tumors. Surgery remains the reference treatment followed more or less by radiotherapy. Recurrences are frequent and the prognosis is reserved for the advanced stage with a modest benefit from chemotherapy. Our case testifies to the interest of multidisciplinary collaboration in the management of this tumour.
References
Penel N, Nisse C, Feddal S, Lartigau E. Épidémiologie des sarcomes des tissus mous de l´adulte. Presse Med. 2001 Oct 6;30(28):1405-13. PubMed | Google Scholar
Eilber FR, Eckardt J. Surgical management of soft tissue sarcoma. Semin Oncol. 1997 Oct;24(5):526-33. PubMed | Google Scholar
Zeren H, Moran CA, Suster S, Fishback NF, Koss MN. Primary pulmonary sarcomas with features of monophasic synovialsarcoma: a clinicopathological, immunohistochemical and ultrastructural study of 25 cases. Hum Pathol. 1995 May;26(5):474-80. PubMed | Google Scholar
Etienne-Mastroianni B, Falchero L, Chalabreysse L, Loire R, Ranchere D, Souquet PJ et al. Primary sarcoma of the lung: a clinicopathologic study of 12 cases. Lung Cancer. 2002 Dec;38(3):283-9. PubMed | Google Scholar
Zahm SH, Fraumeni JF. The epidemiology of soft tissue sarcoma. Semin Oncol. 1997;25(5):504-14. PubMed | Google Scholar
Duran-Mendicuti A, Costello P, Vargas SO. Primary synovial sarcoma of the chest: radiographic and clinicopathologic correlation. J Thorac Imaging. 2003 Apr;18(2):87-93. PubMed | Google Scholar
Eilber Fc, Dry SM. Diagnosis and management of synovial sarcoma. J Surg Oncol. 2008 Mar 15;97(4):314-20. PubMed | Google Scholar
Skytting BT, Bauer HC, Perfekt R, Nilson G, Larson O. Ki 67 is strongly pronostic in synovial sarcoma: Analysis based on 86 patients from the scandinavian sarcoma group register. Br J cancer. 1999 Aug;80(11):1809-14. PubMed | Google Scholar
Authors:
Badache K.
Djaafer M.
Meskouri K.
Medjdoub Y.
Kara Z.
Bendjoudi K.
P: 52-57